LiSiCA Software
LiSiCA Command Line
Download the current version:
- Windows 64: LiSiCAx64.exe
- Windows 32: LiSiCAx86.exe
- Linux: lisica
- Mac: lisicaMAC
- Source: lisica.zip
Example Input Files
- Reference molecule: reference.mol2
- Target molecules: database.mol2
- Acyclovir: acyclovir.mol2
- Aspirin: aspirin.mol2
- Dopamine: dopamine.mol2
- Paxlovid: paxlovid.mol2
- Remdesivir: remdesivir.mol2
Release Notes
1.0.1
- Reads gzipped mol2 files (mol2.gz) for target molecules
Linux: Mark the file as executable:
- chmod +x lisica
Linux from source: Unzip the source code, change the current directory to the 'lisica' folder and compile LiSiCA by typing 'make'.
- unzip lisica.zip
- cd lisica
- make
General Use
- LiSiCA{x86|x64}.exe -R <path to reference molecule> -T <path to target molecules> [parameters]
- ./lisica -R <path to reference molecule> -T <path to target molecules> [parameters]
Input Parameters
-
-n
Description: number of CPU threads to use
Default value: the default is to try to detect the number of CPUs and use all of them or, failing that, use 1 -
-d
Description: product graph dimension
Possible input: 2, 3
Default value: 2 -
-m
Description: maximum allowed atom spatial distance difference for the 3D product graph measured in angstroms
Default value: 1.0 -
-s
Description: maximum allowed shortest path difference for the 2D product graph measured in the number of covalent bonds between atoms
Default value: 1 -
-h
Description: consider hydrogens.
Default value: False -
-w
Description: number of highest ranked molecules to write to output
Default value: 0 -
-c
Description: maximum allowed number of highest scoring conformations to be outputed
Default value: 1 -
--help
Description: print LiSiCA parameters
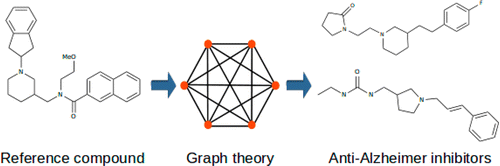
The -d option tells LiSiCA which screening option to use. The 2D option is based on finding similarities between ligands which have the same (-s option = 0) or similar (-s option > 0) number of covalent bond between the same type of atoms. Accordingly, the 3D option is based on finding similarities between ligands where spatial distance between the same type of atoms is similar (or the same if the -m option = 0.0).
The -m option corresponds to the maximum allowed difference in spatial distances between atoms of the two compared product graph vertices. Lesser values correspond to a more rigorous screening. This option can only be used in combination with the -d 3 (3D screening option) option.
The -s option corresponds to the maximum allowed difference in shortest-path lengths between atoms of the two compared product graph vertices. Lesser values correspond to a more rigorous screening. This option can only be used in combination with the -d 2 (2D screening) option.
If the -w option is set to a value higher than 0, LiSiCA will create mol2 files of the highest scoring target molecules with a comment section at the end of the file where the matching atom pairs are displayed.
The -c option corresponds to the maximum number of outputted files of one molecule in different conformations.
The -h option lets the user choose if the hydrogen atoms are to be considered for the calculation of the similarity using the maximum clique algorithm. By default, hydrogen atoms are not considered in finding the largest substructure common to the reference and target molecules, so as to obtain faster results.
Examples
- 1. 2D screening with default options where the final results are saved to the results_2D.dat file:
- ./lisica -R reference.mol2 -T database.mol2[.gz] > results_2D.dat
- 2. 3D screening with default options where the final results are saved to the results_3D.dat:
- ./lisica -R reference.mol2 -T database.mol2[.gz] -d 3 > results_3D.dat
- 3. Rigorous 3D screening where results are send to the standard output only:
- ./lisica -R reference.mol2 -T database.mol2[.gz] -d 3 -m 0.5
- 4. 3D screening where 30 highest scoring target molecules are written in mol2 files with their matching atoms (to the ones in the reference molecule) displayed in the comment section of the mol2 file. Two mol2 files of the best scoring conformers for each molecule are allowed to be written.
- ./lisica -R reference.mol2 -T database.mol2[.gz] -d 3 -w 30 -c 2
Related Publications
 S. Lesnik, T. Stular, B. Brus, D. Knez, S. Gobec, D. Janezic, J. Konc, LiSiCA: A Software for Ligand-Based Virtual Screening and Its Application for the Discovery of Butyrylcholinesterase Inhibitors, J. Chem. Inf. Model., 2015, 55, 1521–1528.
S. Lesnik, T. Stular, B. Brus, D. Knez, S. Gobec, D. Janezic, J. Konc, LiSiCA: A Software for Ligand-Based Virtual Screening and Its Application for the Discovery of Butyrylcholinesterase Inhibitors, J. Chem. Inf. Model., 2015, 55, 1521–1528.
title={LiSiCA: A Software for Ligand-Based Virtual Screening and Its Application for the Discovery of Butyrylcholinesterase Inhibitors},
author={Lešnik, Samo and Štular, Tanja and Brus, Boris and Knez, Damijan and Gobec, Stanislav and Janežič, Dušanka and Konc, Janez},
journal={Journal of chemical information and modeling},
volume={55},
number={8},
pages={1521--1528},
year={2015},
publisher={ACS Publications}
}
%T LiSiCA: A Software for Ligand-Based Virtual Screening and Its Application for the Discovery of Butyrylcholinesterase Inhibitors
%A Lešnik, Samo
%A Štular, Tanja
%A Brus, Boris
%A Knez, Damijan
%A Gobec, Stanislav
%A Janežič, Dušanka
%A Konc, Janez
%J Journal of chemical information and modeling
%V 55
%N 8
%P 1521-1528
%@ 1549-9596
%D 2015
%I ACS Publications
T1 - LiSiCA: A Software for Ligand-Based Virtual Screening and Its Application for the Discovery of Butyrylcholinesterase Inhibitors
A1 - Lešnik, Samo
A1 - Štular, Tanja
A1 - Brus, Boris
A1 - Knez, Damijan
A1 - Gobec, Stanislav
A1 - Janežič, Dušanka
A1 - Konc, Janez
JO - Journal of chemical information and modeling
VL - 55
IS - 8
SP - 1521
EP - 1528
SN - 1549-9596
Y1 - 2015
PB - ACS Publications
ER -